Cystic fibrosis is a disease that causes thick, sticky mucus to develop in the lungs, gastrointestinal tract, and other areas of the body. It is one of the most common chronic lung diseases in children and young adults. It is a dangerous disorder.
What Causes Cystic Fibrosis
Cystic fibrosis is a disease that is given through families. It is triggered by a malfunctioning gene that makes the body produce abnormally thick and sticky fluid, called mucus. This mucus builds up in the breathing passages of the lungs and in the pancreas.
The buildup of mucus results in life-threatening lung infections and major digestion problems. The disease may likewise impact the sweat glands and a man’s reproductive system.
Health Support: This Vitamin K2 + D3 Complex is essential for bone density, cardiovascular health, and immune function. It’s a highly-rated formula for those looking to maintain optimal nutrient levels. You can find it on Amazon.
Many people bring a Cystic fibrosis gene, but do not have symptoms. This is due to the fact that a person with Cystic fibrosis must inherit 2 faulty genes, 1 from each parent. Some white Americans have the Cystic fibrosis gene. It is more common amongst those of northern or main European descent.
A lot of children with Cystic fibrosis are detected by age 2. For a little number, the disease is not spotted up until age 18 or older. These children frequently have a milder kind of the disease.
Symptoms of Cystic Fibrosis
Symptoms in newborns might consist of:
- Delayed development
- Failure to put on weight normally during childhood
- No bowel movements in first 24 to 48 hours of life
- Salty skin
Symptoms associated with bowel function may consist of:
Health Support: This high-absorption Magnesium Glycinate (200 mg) is gentle on the stomach and supports muscle relaxation, better sleep, and metabolic health. You can find this trusted formula on Amazon.
- Belly pain from severe constipation
- Increased gas, bloating, or a belly that appears distended (swollen).
- Nausea and loss of appetite.
- Stools that are pale or clay-colored, nasty smelling, have mucus, or that float.
- Weight-loss.
Symptoms related to the lungs and sinuses might consist of:
- Coughing or increased mucus in the sinuses or lungs.
- Tiredness.
- Nasal congestion caused by nasal polyps.
- Repeated episodes of pneumonia (symptoms of pneumonia in somebody with cystic fibrosis consist of fever, increased coughing and shortness of breath, increased mucus, and loss of appetite).
- Sinus pain or pressure brought on by infection or polyps.
Symptoms that might be seen later in life:
- Infertility (in men).
- Repetitive inflammation of the pancreas (pancreatitis).
- Respiratory symptoms.
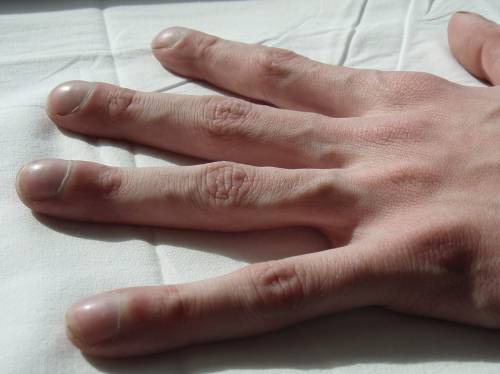
- Clubbed fingers.
How Is Cystic Fibrosis Diagnosed?
A blood test is done to help find Cystic fibrosis. The test searches for changes in the Cystic fibrosis gene. Other tests used to detect Cystic fibrosis consist of:
- Immunoreactive trypsinogen (IRT) test is a standard newborn screening test for Cystic fibrosis. A high level of IRT suggests possible Cystic fibrosis and needs further screening.
- Sweat chloride test is the standard diagnostic test for Cystic fibrosis. A high salt level in the individual’s sweat signifies the disease.
Other tests that determine issues that can be associated with Cystic fibrosis consist of:
- Secretin stimulation test.
- Fecal fat test.
- Trypsin and chymotrypsin in stool.
- Chest x-ray or CT scan.
- Measurement of pancreatic function.
- Lung function tests.
- Upper GI and little bowel series.
Cystic Fibrosis Treatment
An early medical diagnosis of Cystic fibrosis and treatment plan can enhance both survival and quality of life. Follow-up and tracking are extremely important. When possible, care should be gotten at a cystic fibrosis specialized center. When children maturate, they should move to a cystic fibrosis specialty center for adults.
Health Support: The WHOOP 5.0 Activity Tracker offers a comprehensive, 24/7 analysis of your body’s recovery, sleep patterns, daily strain, and key health biometrics. It’s an invaluable tool for anyone looking to optimize their energy, manage stress, and understand what their body truly needs. You can find it on Amazon.
Treatment for lung issues includes:
- Inhaled medications to assist open the respiratory tracts.
- Influenza vaccine and pneumococcal polysaccharide vaccine (PPV) annual (ask your health care service provider).
- Other medications that are offered by a breathing treatment to thin mucus and make it simpler to cough up are
- DNAse enzyme. therapy and highly focused salt options (hypertonic saline).
- Lung transplant is a choice in some cases.
- Antibiotics to avoid and treat lung and sinus infections. They may be taken by mouth, or given in the veins or by breathing treatments. People with Cystic fibrosis may take antibiotics only when needed, or all the time. Dosages are frequently higher than normal.
- Oxygen therapy might be needed as lung disease gets worse.
Lung problems are also treated with therapies to thin the mucus. This makes it simpler to cough the mucus out of the lungs.
These techniques consist of:
- Activity or workout that causes you to breathe deeply.
- Devices that are used during the day to help clear the airways of too much mucus.
- Manual chest percussion (or chest physiotherapy), in which a member of the family or a therapist lightly claps the individual’s chest, back, and area under the arms.
Treatment for bowel and dietary issues may include:
- An unique diet high in protein and calories for older children and adults.
- Pancreatic enzymes to help absorb fats and protein, which are taken with every meal.
- Vitamin supplements, particularly vitamins A, D, E, and K.
- Your service provider can recommend other treatments if you have very difficult stools.
Ivacaftor is a medicine that treats particular types of Cystic fibrosis. It improves the function of one of the defective genes that causes Cystic fibrosis. As an outcome, there is less accumulation of thick mucus in the lungs. Other Cystic fibrosis symptoms are improved also.
Care and keeping an eye on at home must consist of:
- Preventing smoke, dust, dirt, fumes, household chemicals, fireplace smoke, and mold or mildew.
- Offering a lot of fluids, particularly to babies and children in heat, when there is diarrhea or loose stools, or during extra exercise.
- Working out 2 or 3 times each week. Swimming, running, and biking ready choices.
- Clearing or raising mucus or secretions from the respiratory tracts. This should be done 1 to 4 times each day.
- Patients, families, and caretakers should find out about doing chest percussion and postural drain to help keep the respiratory tracts clear.
Benefits of Cystic Fibrosis Support Groups
You can ease the stress of disease by joining a cystic fibrosis support system. Showing others who have common experiences and issues can help your household to not feel alone.
What Is Cystic Fibrosis Life Expectancy?
Many children with Cystic fibrosis stay in health until they maturate. They have the ability to participate in a lot of activities and attend school. Numerous young adults with Cystic fibrosis surface college or discover jobs.
Lung disease ultimately intensifies to the point where the individual is disabled. Today, the typical life expectancy for people with Cystic fibrosis who live to their adult years has to do with 37 years*.
Death is most often triggered by lung complications.
Cystic Fibrosis Possible Complications
The most common complication is chronic respiratory infection.
Other complications consist of:
- Liver disease or liver failure, pancreatitis, biliary cirrhosis.
- Chronic breathing failure.
- Coughing up blood.
- Nasal polyps and sinusitis.
- Infertility.
- Malnutrition.
- Right-sided heart failure (cor pulmonale).
- Pneumothorax.
- Diabetes.
- Pneumonia that keeps coming back.
- Osteoporosis and arthritis.
- Bowel problems, such as gallstones, digestive tract obstruction, and rectal prolapse.
When to Contact a Medical Professional
Call your company if a baby or child has symptoms of Cystic fibrosis, and experiences:
- Fever, increased coughing, changes in sputum or blood in sputum, anorexia nervosa, or other signs of pneumonia.
- Increased weight reduction.
- More frequent defecation or stools that are foul-smelling or have more mucus.
- Swollen belly or increased bloating.
Call your supplier if a person with Cystic fibrosis establishes new symptoms or if symptoms become worse, especially severe breathing problem or coughing up blood.
Cystic Fibrosis Prevention Measures
Cystic fibrosis can not be avoided. Evaluating those with a family history of the disease may identify the Cystic fibrosis gene in numerous providers.
—–
*Life-expectancy prognosis for people with cystic fibrosis based on the analysis of clinical and laboratory data and can vary from person to person.